BGGN 213 Spring 2019 Classwork
https://androidpcguy.github.io/bggn213/
Overview
Today we’ll be going over R functions, but will start off with some file reading first.
plot(1:10, type = 'l', col = 'blue')
test1 file
test1 <- read.table('test1.txt', header = T, sep = ',')
test1
## Col1 Col2 Col3
## 1 1 2 3
## 2 4 5 6
## 3 7 8 9
## 4 a b c
test2 file
test2 <- read.table('./test2.txt', sep = '$', header = T)
test2
## Col1 Col2 Col3
## 1 1 2 3
## 2 4 5 6
## 3 7 8 9
## 4 a b c
test3 file
test3 <- read.table('./test3.txt', header = F)
test3
## V1 V2 V3
## 1 1 6 a
## 2 2 7 b
## 3 3 8 c
## 4 4 9 d
## 5 5 10 e
First function
add <- function(x, y = 1) {
# the body
x + y
}
add(c(1, 3, 5))
## [1] 2 4 6
# add(1, 3, 5)
# add(x = 1, y = "barry")
rescale <- function(x) {
rng <- range(x)
(x - rng[1]) / (rng[2] - rng[1])
}
rescale2 <- function(x, na.rm = T) {
rng <- range(x, na.rm = na.rm)
(x - rng[1]) / (rng[2] - rng[1])
}
x <- c(1:10, NA)
rng <- range(x)
(x - rng[1]) / (rng[2] - rng[1])
## [1] NA NA NA NA NA NA NA NA NA NA NA
rng
## [1] NA NA
print(rescale2(c(1:10, NA)))
## [1] 0.0000000 0.1111111 0.2222222 0.3333333 0.4444444 0.5555556 0.6666667
## [8] 0.7777778 0.8888889 1.0000000 NA
print(rescale2(c(1:10, NA), na.rm = F))
## [1] NA NA NA NA NA NA NA NA NA NA NA
Another example extension…
rescale3 <- function(x, na.rm=TRUE, plot=FALSE) {
rng <-range(x, na.rm=na.rm)
print("Hello")
answer <- (x - rng[1]) / (rng[2] - rng[1])
print("is it me you are looking for?")
if(plot) {
plot(answer, typ="b", lwd=4)
print("Don't sing please!!!!")
}
print("I can see it in ...")
return(answer)
}
rescale3(1:10)
## [1] "Hello"
## [1] "is it me you are looking for?"
## [1] "I can see it in ..."
## [1] 0.0000000 0.1111111 0.2222222 0.3333333 0.4444444 0.5555556 0.6666667
## [8] 0.7777778 0.8888889 1.0000000
rescale3(1:10, plot = T)
## [1] "Hello"
## [1] "is it me you are looking for?"
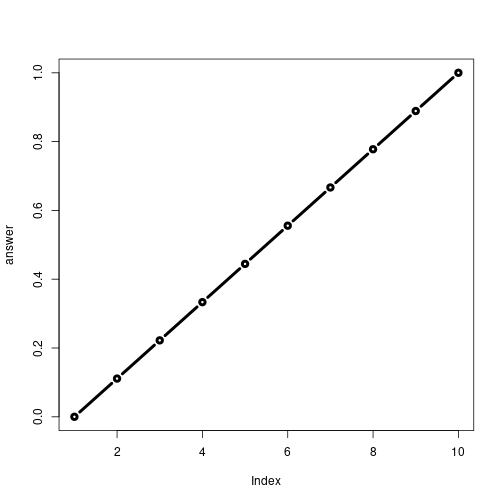
## [1] "Don't sing please!!!!"
## [1] "I can see it in ..."
## [1] 0.0000000 0.1111111 0.2222222 0.3333333 0.4444444 0.5555556 0.6666667
## [8] 0.7777778 0.8888889 1.0000000
Practical protein interaction example
source('./bio3d_example.R', echo = T)
##
## > require(bio3d)
## Loading required package: bio3d
##
## > prot_interact <- function(pdb.name, chain = "A", elety = "CA",
## + ret = F) {
## + pdb.dat <- read.pdb(pdb.name)
## + chain <- trim.pdb(pdb.dat .... [TRUNCATED]
prot_interact
## function (pdb.name, chain = "A", elety = "CA", ret = F)
## {
## pdb.dat <- read.pdb(pdb.name)
## chain <- trim.pdb(pdb.dat, chain = chain, elety = elety)
## chain.b <- chain$atom$b
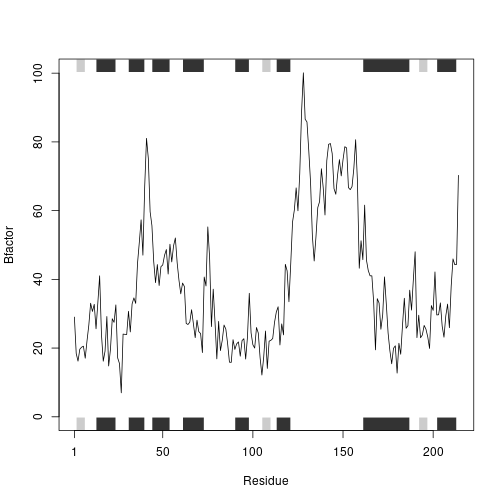
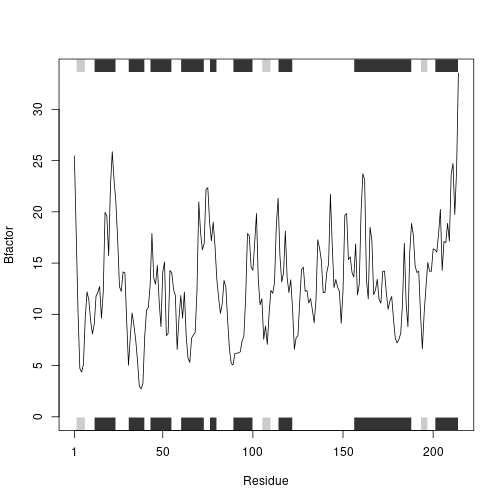
## plotb3(chain.b, sse = chain, typ = "l", ylab = "Bfactor")
## if (ret) {
## return(chain.b)
## }
## }
int1 <- prot_interact('4AKE', ret = T)
## Note: Accessing on-line PDB file
int2 <- prot_interact('1AKE', ret = T)
## Note: Accessing on-line PDB file
## PDB has ALT records, taking A only, rm.alt=TRUE
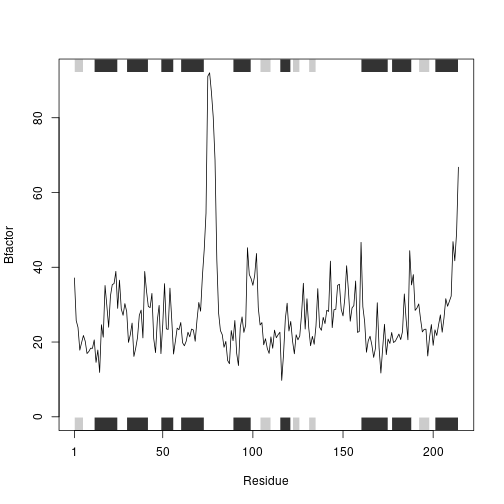
int3 <- prot_interact('1E4Y', ret = T)
## Note: Accessing on-line PDB file
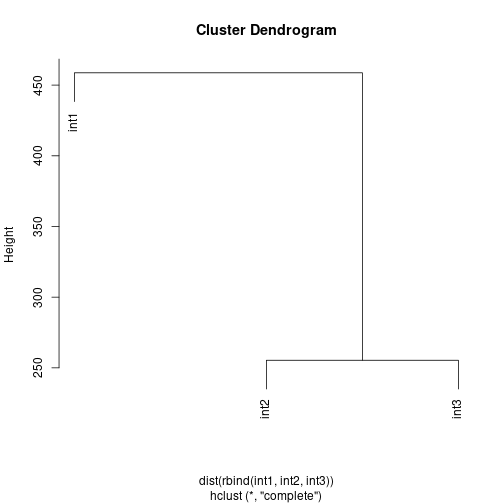
hc <- hclust( dist( rbind(int1, int2, int3) ) )
plot(hc)