BGGN 213 Spring 2019 Classwork
https://androidpcguy.github.io/bggn213/
R Notebook
Revisit the PDB
Downloaded csv file from www.rcsb.org/stats/summary on 2019-05-08
db <- read.csv('pdb_stats.csv', row.names = 1)
db
## Proteins Nucleic.Acids Protein.NA.Complex Other Total
## X-Ray 127027 2012 6549 8 135596
## NMR 11064 1279 259 8 12610
## Electron Microscopy 2296 31 803 0 3130
## Other 256 4 6 13 279
## Multi Method 131 5 2 1 139
Question 1: How many structures solved by X-ray and EM?
xray <- db$Total[1]
em <- db$Total[3]
total <- sum(db$Total)
print(paste(xray/total * 100, "% of structures solved by xray and", em/total*100, "% of structures solved by EM"))
## [1] "89.3525047115727 % of structures solved by xray and 2.0625485983895 % of structures solved by EM"
print(paste(sum(db$Proteins)/total*100, "% are protein structures and", sum(db$Nucleic.Acids)/total*100, "% are nucleic acid structures."))
## [1] "92.7646058752982 % are protein structures and 2.19499980231164 % are nucleic acid structures."
There are 1157 HIV1-protease structures in PDB.
Section 2: VMD
Hydrogens are not visible in this particular pdb file
Section 3. Using Bio3D
library(bio3d)
Reading in the HIV PDB file…
pdb <- read.pdb('1hsg.pdb')
pdb
##
## Call: read.pdb(file = "1hsg.pdb")
##
## Total Models#: 1
## Total Atoms#: 1686, XYZs#: 5058 Chains#: 2 (values: A B)
##
## Protein Atoms#: 1514 (residues/Calpha atoms#: 198)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 172 (residues: 128)
## Non-protein/nucleic resid values: [ HOH (127), MK1 (1) ]
##
## Protein sequence:
## PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
## QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
## ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
## VNIIGRNLLTQIGCTLNF
##
## + attr: atom, xyz, seqres, helix, sheet,
## calpha, remark, call
# one letter code aa321() function
aa321(pdb$seqres)
## [1] "P" "Q" "I" "T" "L" "W" "Q" "R" "P" "L" "V" "T" "I" "K" "I" "G" "G"
## [18] "Q" "L" "K" "E" "A" "L" "L" "D" "T" "G" "A" "D" "D" "T" "V" "L" "E"
## [35] "E" "M" "S" "L" "P" "G" "R" "W" "K" "P" "K" "M" "I" "G" "G" "I" "G"
## [52] "G" "F" "I" "K" "V" "R" "Q" "Y" "D" "Q" "I" "L" "I" "E" "I" "C" "G"
## [69] "H" "K" "A" "I" "G" "T" "V" "L" "V" "G" "P" "T" "P" "V" "N" "I" "I"
## [86] "G" "R" "N" "L" "L" "T" "Q" "I" "G" "C" "T" "L" "N" "F" "P" "Q" "I"
## [103] "T" "L" "W" "Q" "R" "P" "L" "V" "T" "I" "K" "I" "G" "G" "Q" "L" "K"
## [120] "E" "A" "L" "L" "D" "T" "G" "A" "D" "D" "T" "V" "L" "E" "E" "M" "S"
## [137] "L" "P" "G" "R" "W" "K" "P" "K" "M" "I" "G" "G" "I" "G" "G" "F" "I"
## [154] "K" "V" "R" "Q" "Y" "D" "Q" "I" "L" "I" "E" "I" "C" "G" "H" "K" "A"
## [171] "I" "G" "T" "V" "L" "V" "G" "P" "T" "P" "V" "N" "I" "I" "G" "R" "N"
## [188] "L" "L" "T" "Q" "I" "G" "C" "T" "L" "N" "F"
# pdb$atom is a dataframe
head(pdb$atom)
## type eleno elety alt resid chain resno insert x y z o
## 1 ATOM 1 N <NA> PRO A 1 <NA> 29.361 39.686 5.862 1
## 2 ATOM 2 CA <NA> PRO A 1 <NA> 30.307 38.663 5.319 1
## 3 ATOM 3 C <NA> PRO A 1 <NA> 29.760 38.071 4.022 1
## 4 ATOM 4 O <NA> PRO A 1 <NA> 28.600 38.302 3.676 1
## 5 ATOM 5 CB <NA> PRO A 1 <NA> 30.508 37.541 6.342 1
## 6 ATOM 6 CG <NA> PRO A 1 <NA> 29.296 37.591 7.162 1
## b segid elesy charge
## 1 38.10 <NA> N <NA>
## 2 40.62 <NA> C <NA>
## 3 42.64 <NA> C <NA>
## 4 43.40 <NA> O <NA>
## 5 37.87 <NA> C <NA>
## 6 38.40 <NA> C <NA>
pdb$atom is a dataframe
Atom selection examples
Let’s select residue 10
inds <- atom.select(pdb, resno = 10)
pdb$atom[inds$atom,]
## type eleno elety alt resid chain resno insert x y z o
## 81 ATOM 81 N <NA> LEU A 10 <NA> 25.905 28.285 9.330 1
## 82 ATOM 82 CA <NA> LEU A 10 <NA> 25.653 28.510 10.750 1
## 83 ATOM 83 C <NA> LEU A 10 <NA> 26.383 29.770 11.208 1
## 84 ATOM 84 O <NA> LEU A 10 <NA> 27.567 29.927 10.938 1
## 85 ATOM 85 CB <NA> LEU A 10 <NA> 26.120 27.284 11.573 1
## 86 ATOM 86 CG <NA> LEU A 10 <NA> 25.161 26.082 11.544 1
## 87 ATOM 87 CD1 <NA> LEU A 10 <NA> 25.895 24.743 11.662 1
## 88 ATOM 88 CD2 <NA> LEU A 10 <NA> 24.206 26.196 12.696 1
## 838 ATOM 839 N <NA> LEU B 10 <NA> 12.134 31.727 -5.504 1
## 839 ATOM 840 CA <NA> LEU B 10 <NA> 11.816 30.740 -6.534 1
## 840 ATOM 841 C <NA> LEU B 10 <NA> 12.459 31.075 -7.877 1
## 841 ATOM 842 O <NA> LEU B 10 <NA> 12.274 32.150 -8.406 1
## 842 ATOM 843 CB <NA> LEU B 10 <NA> 10.303 30.637 -6.738 1
## 843 ATOM 844 CG <NA> LEU B 10 <NA> 9.483 30.307 -5.497 1
## 844 ATOM 845 CD1 <NA> LEU B 10 <NA> 8.028 30.334 -5.876 1
## 845 ATOM 846 CD2 <NA> LEU B 10 <NA> 9.845 28.975 -4.951 1
## b segid elesy charge
## 81 28.83 <NA> N <NA>
## 82 31.57 <NA> C <NA>
## 83 30.48 <NA> C <NA>
## 84 31.00 <NA> O <NA>
## 85 31.09 <NA> C <NA>
## 86 35.91 <NA> C <NA>
## 87 40.15 <NA> C <NA>
## 88 40.51 <NA> C <NA>
## 838 18.74 <NA> N <NA>
## 839 24.75 <NA> C <NA>
## 840 28.33 <NA> C <NA>
## 841 34.15 <NA> O <NA>
## 842 22.30 <NA> C <NA>
## 843 26.19 <NA> C <NA>
## 844 26.68 <NA> C <NA>
## 845 25.72 <NA> C <NA>
atom.select(pdb, resno = 10, value = T)
##
## Call: trim.pdb(pdb = pdb, sele)
##
## Total Models#: 1
## Total Atoms#: 16, XYZs#: 48 Chains#: 2 (values: A B)
##
## Protein Atoms#: 16 (residues/Calpha atoms#: 2)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 0 (residues: 0)
## Non-protein/nucleic resid values: [ none ]
##
## Protein sequence:
## LL
##
## + attr: atom, helix, sheet, seqres, xyz,
## calpha, call
We want to select only the protein sequence without water or the drug already there. We also want to isolate the ligand itself.
protein.only <- atom.select(pdb, string = 'protein', value = TRUE)
ligand.only <- atom.select(pdb, string = 'ligand', value = TRUE)
protein.only
##
## Call: trim.pdb(pdb = pdb, sele)
##
## Total Models#: 1
## Total Atoms#: 1514, XYZs#: 4542 Chains#: 2 (values: A B)
##
## Protein Atoms#: 1514 (residues/Calpha atoms#: 198)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 0 (residues: 0)
## Non-protein/nucleic resid values: [ none ]
##
## Protein sequence:
## PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
## QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
## ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
## VNIIGRNLLTQIGCTLNF
##
## + attr: atom, helix, sheet, seqres, xyz,
## calpha, call
ligand.only
##
## Call: trim.pdb(pdb = pdb, sele)
##
## Total Models#: 1
## Total Atoms#: 45, XYZs#: 135 Chains#: 1 (values: B)
##
## Protein Atoms#: 0 (residues/Calpha atoms#: 0)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 45 (residues: 1)
## Non-protein/nucleic resid values: [ MK1 (1) ]
##
## + attr: atom, helix, sheet, seqres, xyz,
## calpha, call
# write the protein and ligand to separate files
write.pdb(pdb = protein.only, file = '1hsg_protein.pdb')
write.pdb(pdb = ligand.only, file = '1hsg_ligand.pdb')
require(bio3d.view)
## Loading required package: bio3d.view
view(protein.only)
## Computing connectivity from coordinates...
Section 6. Multiple sequence alignment
# Download some example PDB files
ids <- c("1TND_B","1AGR_A","1TAG_A","1GG2_A","1KJY_A","4G5Q_A")
files <- get.pdb(ids, split = TRUE)
## Warning in get.pdb(ids, split = TRUE): ./1TND.pdb exists. Skipping download
## Warning in get.pdb(ids, split = TRUE): ./1AGR.pdb exists. Skipping download
## Warning in get.pdb(ids, split = TRUE): ./1TAG.pdb exists. Skipping download
## Warning in get.pdb(ids, split = TRUE): ./1GG2.pdb exists. Skipping download
## Warning in get.pdb(ids, split = TRUE): ./1KJY.pdb exists. Skipping download
## Warning in get.pdb(ids, split = TRUE): ./4G5Q.pdb exists. Skipping download
##
|
| | 0%
|
|=========== | 17%
|
|====================== | 33%
|
|================================ | 50%
|
|=========================================== | 67%
|
|====================================================== | 83%
|
|=================================================================| 100%
# Extract and align the chains we are interested in
pdbs <- pdbaln(files, fit = TRUE)
## Reading PDB files:
## ./split_chain/1TND_B.pdb
## ./split_chain/1AGR_A.pdb
## ./split_chain/1TAG_A.pdb
## ./split_chain/1GG2_A.pdb
## ./split_chain/1KJY_A.pdb
## ./split_chain/4G5Q_A.pdb
## ..... PDB has ALT records, taking A only, rm.alt=TRUE
## .
##
## Extracting sequences
##
## pdb/seq: 1 name: ./split_chain/1TND_B.pdb
## pdb/seq: 2 name: ./split_chain/1AGR_A.pdb
## pdb/seq: 3 name: ./split_chain/1TAG_A.pdb
## pdb/seq: 4 name: ./split_chain/1GG2_A.pdb
## pdb/seq: 5 name: ./split_chain/1KJY_A.pdb
## pdb/seq: 6 name: ./split_chain/4G5Q_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
# Print to screen a summary of the 'pdbs' object
pdbs
## 1 . . . . 50
## [Truncated_Name:1]1TND_B.pdb --------------------------ARTVKLLLLGAGESGKSTIVKQMK
## [Truncated_Name:2]1AGR_A.pdb LSAEDKAAVERSKMIDRNLREDGEKAAREVKLLLLGAGESGKSTIVKQMK
## [Truncated_Name:3]1TAG_A.pdb --------------------------ARTVKLLLLGAGESGKSTIVKQMK
## [Truncated_Name:4]1GG2_A.pdb LSAEDKAAVERSKMIDRNLREDGEKAAREVKLLLLGAGESGKSTIVKQMK
## [Truncated_Name:5]1KJY_A.pdb -------------------------GAREVKLLLLGAGESGKSTIVKQMK
## [Truncated_Name:6]4G5Q_A.pdb --------------------------AREVKLLLLGAGESGKSTIVKQMK
## ** *********************
## 1 . . . . 50
##
## 51 . . . . 100
## [Truncated_Name:1]1TND_B.pdb IIHQDGYSLEECLEFIAIIYGNTLQSILAIVRAMTTLNIQYGDSARQDDA
## [Truncated_Name:2]1AGR_A.pdb IIHEAGYSEEECKQYKAVVYSNTIQSIIAIIRAMGRLKIDFGDAARADDA
## [Truncated_Name:3]1TAG_A.pdb IIHQDGYSLEECLEFIAIIYGNTLQSILAIVRAMTTLNIQYGDSARQDDA
## [Truncated_Name:4]1GG2_A.pdb IIHEAGYSEEECKQYKAVVYSNTIQSIIAIIRAMGRLKIDFGDAARADDA
## [Truncated_Name:5]1KJY_A.pdb IIHEAGYSEEECKQYKAVVYSNTIQSIIAIIRAMGRLKIDFGDSARADDA
## [Truncated_Name:6]4G5Q_A.pdb IIHEAGYSEEECKQYKAVVYSNTIQSIIAIIRAMGRLKIDFGDSARADDA
## *** *** *** ^ *^^* **^***^**^*** * * ^** ** ***
## 51 . . . . 100
##
## 101 . . . . 150
## [Truncated_Name:1]1TND_B.pdb RKLMHMADTIEEGTMPKEMSDIIQRLWKDSGIQACFDRASEYQLNDSAGY
## [Truncated_Name:2]1AGR_A.pdb RQLFVLAGAAEEGFMTAELAGVIKRLWKDSGVQACFNRSREYQLNDSAAY
## [Truncated_Name:3]1TAG_A.pdb RKLMHMADTIEEGTMPKEMSDIIQRLWKDSGIQACFDRASEYQLNDSAGY
## [Truncated_Name:4]1GG2_A.pdb RQLFVLAGAAEEGFMTAELAGVIKRLWKDSGVQACFNRSREYQLNDSAAY
## [Truncated_Name:5]1KJY_A.pdb RQLFVLAGAAEEGFMTAELAGVIKRLWKDSGVQACFNRSREYQLNDSAAY
## [Truncated_Name:6]4G5Q_A.pdb RQLFVLAGAAEEGFMTAELAGVIKRLWKDSGVQACFNRSREYQLNDSAAY
## * * ^* *** * *^ ^* *******^**** * ********^*
## 101 . . . . 150
##
## 151 . . . . 200
## [Truncated_Name:1]1TND_B.pdb YLSDLERLVTPGYVPTEQDVLRSRVKTTGIIETQFSFKDLNFRMFDVGGQ
## [Truncated_Name:2]1AGR_A.pdb YLNDLDRIAQPNYIPTQQDVLRTRVKTTGIVETHFTFKDLHFKMFDVGGQ
## [Truncated_Name:3]1TAG_A.pdb YLSDLERLVTPGYVPTEQDVLRSRVKTTGIIETQFSFKDLNFRMFDVGGQ
## [Truncated_Name:4]1GG2_A.pdb YLNDLDRIAQPNYIPTQQDVLRTRVKTTGIVETHFTFKDLHFKMFDVGAQ
## [Truncated_Name:5]1KJY_A.pdb YLNDLDRIAQPNYIPTQQDVLRTRVKTTGIVETHFTFKDLHFKMFDVGGQ
## [Truncated_Name:6]4G5Q_A.pdb YLNDLDRIAQPNYIPTQQDVLRTRVKTTGIVETHFTFKDLHFKMFDVGGQ
## ** **^*^ * *^** *****^*******^** *^**** *^*****^*
## 151 . . . . 200
##
## 201 . . . . 250
## [Truncated_Name:1]1TND_B.pdb RSERKKWIHCFEGVTCIIFIAALSAYDMVLVEDDEVNRMHESLHLFNSIC
## [Truncated_Name:2]1AGR_A.pdb RSERKKWIHCFEGVTAIIFCVALSDYDLVLAEDEEMNRMHESMKLFDSIC
## [Truncated_Name:3]1TAG_A.pdb RSERKKWIHCFEGVTCIIFIAALSAYDMVLVEDDEVNRMHESLHLFNSIC
## [Truncated_Name:4]1GG2_A.pdb RSERKKWIHCFEGVTAIIFCVALSDYDLVLAEDEEMNRMHESMKLFDSIC
## [Truncated_Name:5]1KJY_A.pdb RSERKKWIHCFEGVTAIIFCVALSDYDLVLAEDEEMNRMHESMKLFDSIC
## [Truncated_Name:6]4G5Q_A.pdb RSERKKWIHCFEGVTAIIFCVALSDYDLVLAEDEEMNRMHESMKLFDSIC
## *************** *** *** **^** **^*^******^^** ***
## 201 . . . . 250
##
## 251 . . . . 300
## [Truncated_Name:1]1TND_B.pdb NHRYFATTSIVLFLNKKDVFSEKIKKAHLSICFPDYNGPNTYEDAGNYIK
## [Truncated_Name:2]1AGR_A.pdb NNKWFTDTSIILFLNKKDLFEEKIKKSPLTICYPEYAGSNTYEEAAAYIQ
## [Truncated_Name:3]1TAG_A.pdb NHRYFATTSIVLFLNKKDVFSEKIKKAHLSICFPDYNGPNTYEDAGNYIK
## [Truncated_Name:4]1GG2_A.pdb NNKWFTDTSIILFLNKKDLFEEKIKKSPLTICYPEYAGSNTYEEAAAYIQ
## [Truncated_Name:5]1KJY_A.pdb NNKWFTDTSIILFLNKKDLFEEKIKKSPLTICYPEYAGSNTYEEAAAYIQ
## [Truncated_Name:6]4G5Q_A.pdb NNKWFTDTSIILFLNKKDLFEEKIKKSPLTICYPEYAGSNTYEEAAAYIQ
## * ^^* ***^*******^* ***** *^**^*^* * ****^*^ **
## 251 . . . . 300
##
## 301 . . . . 350
## [Truncated_Name:1]1TND_B.pdb VQFLELNMRRDVKEIYSHMTCATDTQNVKFVFDAVTDIIIKE--------
## [Truncated_Name:2]1AGR_A.pdb CQFEDLNKRKDTKEIYTHFTCATDTKNVQFVFDAVTDVIIKNNLKDCGLF
## [Truncated_Name:3]1TAG_A.pdb VQFLELNMRRDVKEIYSHMTCATDTQNVKFVFDAVTDIII----------
## [Truncated_Name:4]1GG2_A.pdb CQFEDLNKRKDTKEIYTHFTCATDTKNVQFVFDAVTDVIIKNNL------
## [Truncated_Name:5]1KJY_A.pdb CQFEDLNKRKDTKEIYTHFTCATDTKNVQFVFDAVTDVIIKNNLK-----
## [Truncated_Name:6]4G5Q_A.pdb CQFEDLNKRKDTKEIYTHFTCATDTKNVQFVFDAVTDVIIKNNLKD----
## ** ^** *^* ****^* ****** ** ********^**
## 301 . . . . 350
##
## Call:
## pdbaln(files = files, fit = TRUE)
##
## Class:
## pdbs, fasta
##
## Alignment dimensions:
## 6 sequence rows; 350 position columns (314 non-gap, 36 gap)
##
## + attr: xyz, resno, b, chain, id, ali, resid, sse, call
rmsd is measuring root mean square distance. We are aligning the
sequences to each other to minimize distance between them.
Basic structure analysis
seqidentity(pdbs)
## ./split_chain/1TND_B.pdb ./split_chain/1AGR_A.pdb
## ./split_chain/1TND_B.pdb 1.000 0.693
## ./split_chain/1AGR_A.pdb 0.693 1.000
## ./split_chain/1TAG_A.pdb 1.000 0.694
## ./split_chain/1GG2_A.pdb 0.690 0.997
## ./split_chain/1KJY_A.pdb 0.696 0.994
## ./split_chain/4G5Q_A.pdb 0.696 0.997
## ./split_chain/1TAG_A.pdb ./split_chain/1GG2_A.pdb
## ./split_chain/1TND_B.pdb 1.000 0.690
## ./split_chain/1AGR_A.pdb 0.694 0.997
## ./split_chain/1TAG_A.pdb 1.000 0.691
## ./split_chain/1GG2_A.pdb 0.691 1.000
## ./split_chain/1KJY_A.pdb 0.697 0.991
## ./split_chain/4G5Q_A.pdb 0.697 0.994
## ./split_chain/1KJY_A.pdb ./split_chain/4G5Q_A.pdb
## ./split_chain/1TND_B.pdb 0.696 0.696
## ./split_chain/1AGR_A.pdb 0.994 0.997
## ./split_chain/1TAG_A.pdb 0.697 0.697
## ./split_chain/1GG2_A.pdb 0.991 0.994
## ./split_chain/1KJY_A.pdb 1.000 1.000
## ./split_chain/4G5Q_A.pdb 1.000 1.000
rmsd(pdbs)
## Warning in rmsd(pdbs): No indices provided, using the 314 non NA positions
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 0.000 1.042 1.281 1.651 2.098 2.367
## [2,] 1.042 0.000 1.628 1.811 1.949 2.244
## [3,] 1.281 1.628 0.000 1.730 1.840 1.885
## [4,] 1.651 1.811 1.730 0.000 1.901 2.032
## [5,] 2.098 1.949 1.840 1.901 0.000 1.225
## [6,] 2.367 2.244 1.885 2.032 1.225 0.000
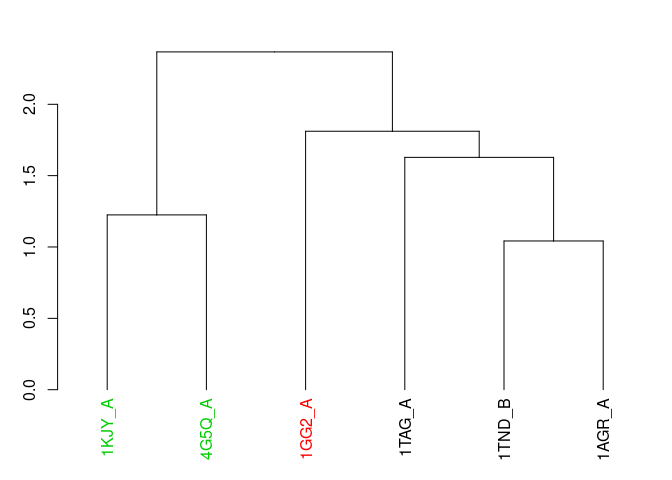
Plot heirarchical clustering results…
distance <- rmsd(pdbs)
## Warning in rmsd(pdbs): No indices provided, using the 314 non NA positions
# clustering
hc <- hclust(as.dist(distance))
grps <- cutree(hc, k = 3)
hc$labels <- ids # set the names properly
# plot the results
hclustplot(hc, k = 3)
PCA
pc.xray <- pca(pdbs)
plot(pc.xray)
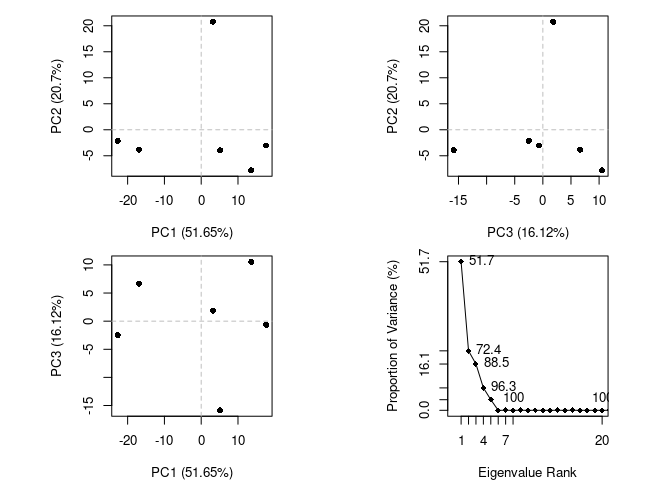
What is PC1 representing???
pc1 <- mktrj(pc.xray, pc = 1, file = 'pc_1.pdb')
view(pc1)
## Potential all C-alpha atom structure(s) detected: Using calpha.connectivity()