BGGN 213 Spring 2019 Classwork
https://androidpcguy.github.io/bggn213/
Class 12
Obtain and prepare our structure for docking
Downloading HIV-pr structure from PDB….
require(bio3d)
## Loading required package: bio3d
prot.id <- '1hsg'
file <- get.pdb(prot.id)
## Warning in get.pdb(prot.id): ./1hsg.pdb exists. Skipping download
pdb <- read.pdb(file)
pdb
##
## Call: read.pdb(file = file)
##
## Total Models#: 1
## Total Atoms#: 1686, XYZs#: 5058 Chains#: 2 (values: A B)
##
## Protein Atoms#: 1514 (residues/Calpha atoms#: 198)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 172 (residues: 128)
## Non-protein/nucleic resid values: [ HOH (127), MK1 (1) ]
##
## Protein sequence:
## PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
## QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
## ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
## VNIIGRNLLTQIGCTLNF
##
## + attr: atom, xyz, seqres, helix, sheet,
## calpha, remark, call
Extract the protein and ligands separately…
pdb.prot <- trim.pdb(pdb, 'protein')
pdb.lig <- trim.pdb(pdb, 'ligand')
pdb.prot
##
## Call: trim.pdb(pdb = pdb, "protein")
##
## Total Models#: 1
## Total Atoms#: 1514, XYZs#: 4542 Chains#: 2 (values: A B)
##
## Protein Atoms#: 1514 (residues/Calpha atoms#: 198)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 0 (residues: 0)
## Non-protein/nucleic resid values: [ none ]
##
## Protein sequence:
## PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
## QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
## ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
## VNIIGRNLLTQIGCTLNF
##
## + attr: atom, helix, sheet, seqres, xyz,
## calpha, call
pdb.lig
##
## Call: trim.pdb(pdb = pdb, "ligand")
##
## Total Models#: 1
## Total Atoms#: 45, XYZs#: 135 Chains#: 1 (values: B)
##
## Protein Atoms#: 0 (residues/Calpha atoms#: 0)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 45 (residues: 1)
## Non-protein/nucleic resid values: [ MK1 (1) ]
##
## + attr: atom, helix, sheet, seqres, xyz,
## calpha, call
# write the pdb files to disk
write.pdb(pdb.prot, '1hsg_protein.pdb')
write.pdb(pdb.lig, '1hsg_ligand.pdb')
Add hydrogens and charges in ADT
We loaded the protein into autodock tools added, hydrogens, and extracted charges on each atom.
Run docking
Run docking with Autodock vina…
vina --cpu 3 --config config.txt --log vina.log
Inspect docking results
We will process the docking results in R
dock.res <- read.pdb('1hsg_ligand_out.pdbqt', multi = TRUE)
write.pdb(dock.res, 'results.pdb')
Docking looks okay, the ligand and new molecule overlap in the protein pocket.
Quantify the docking results
orig <- read.pdb('1hsg_ligand.pdbqt')
rmsd(orig, dock.res)
## [1] 10.902 6.198 4.373 11.876 9.829 9.564 10.220 12.554 9.797 9.620
## [11] 9.636 11.050 9.578 9.380 11.140 10.774
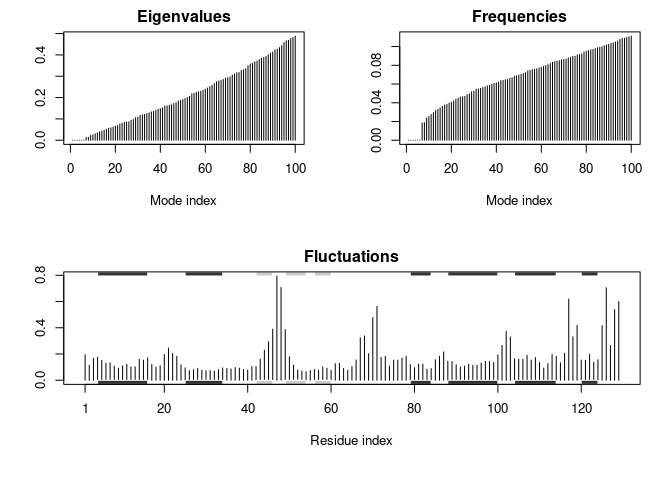
Normal Mode Analysis (NMA)
We’ll do NMA now…
pdb <- read.pdb('1hel')
## Note: Accessing on-line PDB file
modes <- nma(pdb)
## Building Hessian... Done in 0.021 seconds.
## Diagonalizing Hessian... Done in 0.11 seconds.
plot(modes, sse = pdb)
mktrj(modes, mode = 7, file = 'nma_7.pdb')