BGGN 213 Spring 2019 Classwork
https://androidpcguy.github.io/bggn213/
Class 14: RNAseq analysis
Akshara Balachandra 5/17/2019
Library imports…
library(DESeq2)
## Loading required package: S4Vectors
## Loading required package: stats4
## Loading required package: BiocGenerics
## Loading required package: parallel
##
## Attaching package: 'BiocGenerics'
## The following objects are masked from 'package:parallel':
##
## clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
## clusterExport, clusterMap, parApply, parCapply, parLapply,
## parLapplyLB, parRapply, parSapply, parSapplyLB
## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs
## The following objects are masked from 'package:base':
##
## anyDuplicated, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter,
## Find, get, grep, grepl, intersect, is.unsorted, lapply, Map,
## mapply, match, mget, order, paste, pmax, pmax.int, pmin,
## pmin.int, Position, rank, rbind, Reduce, rownames, sapply,
## setdiff, sort, table, tapply, union, unique, unsplit, which,
## which.max, which.min
##
## Attaching package: 'S4Vectors'
## The following object is masked from 'package:base':
##
## expand.grid
## Loading required package: IRanges
## Loading required package: GenomicRanges
## Loading required package: GenomeInfoDb
## Loading required package: SummarizedExperiment
## Loading required package: Biobase
## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.
## Loading required package: DelayedArray
## Loading required package: matrixStats
##
## Attaching package: 'matrixStats'
## The following objects are masked from 'package:Biobase':
##
## anyMissing, rowMedians
## Loading required package: BiocParallel
##
## Attaching package: 'DelayedArray'
## The following objects are masked from 'package:matrixStats':
##
## colMaxs, colMins, colRanges, rowMaxs, rowMins, rowRanges
## The following objects are masked from 'package:base':
##
## aperm, apply, rowsum
## Registered S3 methods overwritten by 'ggplot2':
## method from
## [.quosures rlang
## c.quosures rlang
## print.quosures rlang
library(tibble)
library(dplyr)
##
## Attaching package: 'dplyr'
## The following object is masked from 'package:matrixStats':
##
## count
## The following object is masked from 'package:Biobase':
##
## combine
## The following objects are masked from 'package:GenomicRanges':
##
## intersect, setdiff, union
## The following object is masked from 'package:GenomeInfoDb':
##
## intersect
## The following objects are masked from 'package:IRanges':
##
## collapse, desc, intersect, setdiff, slice, union
## The following objects are masked from 'package:S4Vectors':
##
## first, intersect, rename, setdiff, setequal, union
## The following objects are masked from 'package:BiocGenerics':
##
## combine, intersect, setdiff, union
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
Read count data and metadata…
counts <- read.csv('data/airway_scaledcounts.csv', stringsAsFactors = F, row.names = 1)
metadata <- read.csv('data/airway_metadata.csv', stringsAsFactors = F)
#counts
#metadata
nrow(counts)
## [1] 38694
sum(metadata$dex == 'control')
## [1] 4
Q1: There are 38694 genes in the dataset. Q2: There are 4 control cell lines
Toy differential gene expression
control.cells <- metadata$id[metadata$dex == 'control']
treated.cells <- metadata$id[metadata$dex == 'treated']
control.sub <- subset(counts, select = control.cells)
treated.sub <- subset(counts, select = treated.cells)
control.means <- apply(control.sub, 1, mean)
treated.means <- apply(treated.sub, 1, mean)
control.mean <- sum(control.means)
treated.mean <- sum(treated.means)
mean.counts <- data.frame(control.means, treated.means)
print(paste('Control mean', round(control.mean)))
## [1] "Control mean 23005324"
print(paste('Treated mean', round(treated.mean)))
## [1] "Treated mean 22196524"
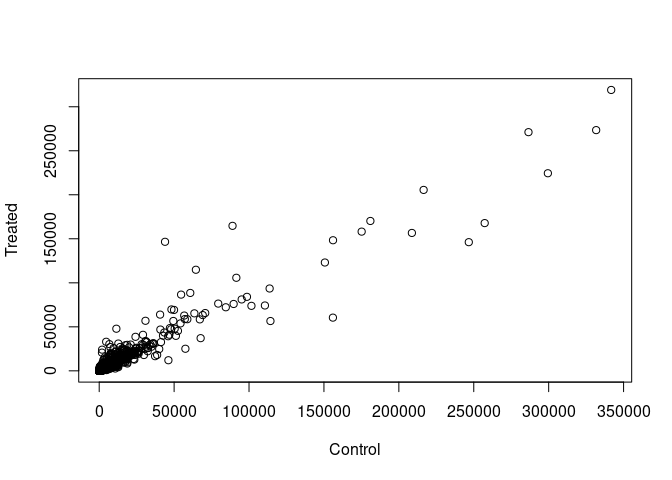
Scatter plot of means of controls vs treated…
plot(control.means, treated.means, xlab = 'Control',
ylab = 'Treated')
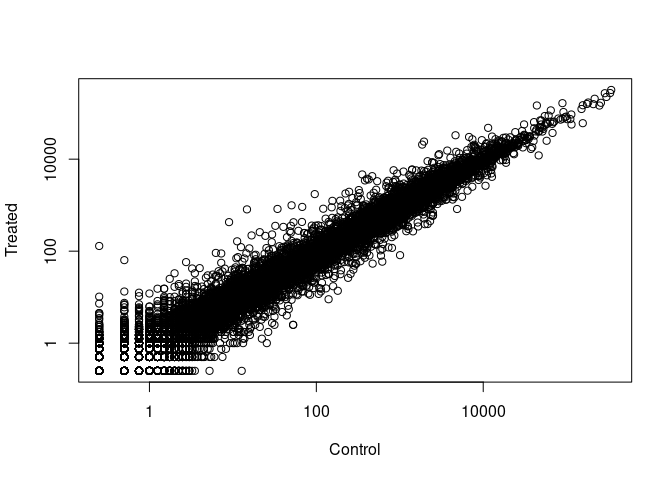
Plot with log scale…
plot(control.means, treated.means, xlab = 'Control',
ylab = 'Treated', log = 'xy')
## Warning in xy.coords(x, y, xlabel, ylabel, log): 15032 x values <= 0
## omitted from logarithmic plot
## Warning in xy.coords(x, y, xlabel, ylabel, log): 15281 y values <= 0
## omitted from logarithmic plot
Let’s combine the control and treated means and calculate log2 fold change…
mean.counts <- data.frame(control.means, treated.means,
row.names = row.names(counts))
mean.counts$log2foldchange <- log((mean.counts$treated.means/mean.counts$control.means),
base = 2)
head(mean.counts)
## control.means treated.means log2foldchange
## ENSG00000000003 900.75 658.00 -0.45303916
## ENSG00000000005 0.00 0.00 NaN
## ENSG00000000419 520.50 546.00 0.06900279
## ENSG00000000457 339.75 316.50 -0.10226805
## ENSG00000000460 97.25 78.75 -0.30441833
## ENSG00000000938 0.75 0.00 -Inf
Let’s filter out the genes that had 0 expression…
mean.counts <- mean.counts %>% rownames_to_column('gene') %>%
filter(is.finite(log2foldchange)) %>% column_to_rownames('gene')
head(mean.counts)
## control.means treated.means log2foldchange
## ENSG00000000003 900.75 658.00 -0.45303916
## ENSG00000000419 520.50 546.00 0.06900279
## ENSG00000000457 339.75 316.50 -0.10226805
## ENSG00000000460 97.25 78.75 -0.30441833
## ENSG00000000971 5219.00 6687.50 0.35769358
## ENSG00000001036 2327.00 1785.75 -0.38194109
Let’s figure out now how many genes are upregulated and downregulated after adding the treatment
up.regulated <- mean.counts %>% rownames_to_column('gene') %>%
filter(log2foldchange > 2) %>% column_to_rownames('gene')
down.regulated <- mean.counts %>% rownames_to_column('gene') %>%
filter(log2foldchange < -2) %>% column_to_rownames('gene')
head(up.regulated)
## control.means treated.means log2foldchange
## ENSG00000004799 270.50 1429.25 2.401558
## ENSG00000006788 2.75 19.75 2.844349
## ENSG00000008438 0.50 2.75 2.459432
## ENSG00000011677 0.50 2.25 2.169925
## ENSG00000015413 0.50 3.00 2.584963
## ENSG00000015592 0.50 2.25 2.169925
head(down.regulated)
## control.means treated.means log2foldchange
## ENSG00000015520 32.00 6.00 -2.415037
## ENSG00000019186 26.50 1.75 -3.920566
## ENSG00000025423 295.00 54.25 -2.443020
## ENSG00000028277 88.25 22.00 -2.004093
## ENSG00000029559 1.25 0.25 -2.321928
## ENSG00000049246 405.00 93.00 -2.122619
print(paste('There are', nrow(up.regulated), 'upregulated genes relative to control'))
## [1] "There are 250 upregulated genes relative to control"
print(paste('There are', nrow(down.regulated), 'downregulated genes relative to control'))
## [1] "There are 367 downregulated genes relative to control"
Adding annotation data
Merge mean.counts with annotation data…
# read in annotations
annot <- read.csv('data/annotables_grch38.csv',
stringsAsFactors = F,
row.names = NULL)
# filter out duplicate entries
annot <- annot[!duplicated(annot$ensgene),]
row.names(annot) <- NULL
annot <- annot %>% column_to_rownames('ensgene')
head(annot)
## entrez symbol chr start end strand
## ENSG00000000003 7105 TSPAN6 X 100627109 100639991 -1
## ENSG00000000005 64102 TNMD X 100584802 100599885 1
## ENSG00000000419 8813 DPM1 20 50934867 50958555 -1
## ENSG00000000457 57147 SCYL3 1 169849631 169894267 -1
## ENSG00000000460 55732 C1orf112 1 169662007 169854080 1
## ENSG00000000938 2268 FGR 1 27612064 27635277 -1
## biotype
## ENSG00000000003 protein_coding
## ENSG00000000005 protein_coding
## ENSG00000000419 protein_coding
## ENSG00000000457 protein_coding
## ENSG00000000460 protein_coding
## ENSG00000000938 protein_coding
## description
## ENSG00000000003 tetraspanin 6 [Source:HGNC Symbol;Acc:HGNC:11858]
## ENSG00000000005 tenomodulin [Source:HGNC Symbol;Acc:HGNC:17757]
## ENSG00000000419 dolichyl-phosphate mannosyltransferase polypeptide 1, catalytic subunit [Source:HGNC Symbol;Acc:HGNC:3005]
## ENSG00000000457 SCY1-like, kinase-like 3 [Source:HGNC Symbol;Acc:HGNC:19285]
## ENSG00000000460 chromosome 1 open reading frame 112 [Source:HGNC Symbol;Acc:HGNC:25565]
## ENSG00000000938 FGR proto-oncogene, Src family tyrosine kinase [Source:HGNC Symbol;Acc:HGNC:3697]
Merge annotation with colmeans now.
mean.counts.meta <- merge(x = mean.counts,
y = annot,
by = 0) %>% # by = 0 (rownames)
column_to_rownames('Row.names')
head(mean.counts.meta)
## control.means treated.means log2foldchange entrez symbol
## ENSG00000000003 900.75 658.00 -0.45303916 7105 TSPAN6
## ENSG00000000419 520.50 546.00 0.06900279 8813 DPM1
## ENSG00000000457 339.75 316.50 -0.10226805 57147 SCYL3
## ENSG00000000460 97.25 78.75 -0.30441833 55732 C1orf112
## ENSG00000000971 5219.00 6687.50 0.35769358 3075 CFH
## ENSG00000001036 2327.00 1785.75 -0.38194109 2519 FUCA2
## chr start end strand biotype
## ENSG00000000003 X 100627109 100639991 -1 protein_coding
## ENSG00000000419 20 50934867 50958555 -1 protein_coding
## ENSG00000000457 1 169849631 169894267 -1 protein_coding
## ENSG00000000460 1 169662007 169854080 1 protein_coding
## ENSG00000000971 1 196651878 196747504 1 protein_coding
## ENSG00000001036 6 143494811 143511690 -1 protein_coding
## description
## ENSG00000000003 tetraspanin 6 [Source:HGNC Symbol;Acc:HGNC:11858]
## ENSG00000000419 dolichyl-phosphate mannosyltransferase polypeptide 1, catalytic subunit [Source:HGNC Symbol;Acc:HGNC:3005]
## ENSG00000000457 SCY1-like, kinase-like 3 [Source:HGNC Symbol;Acc:HGNC:19285]
## ENSG00000000460 chromosome 1 open reading frame 112 [Source:HGNC Symbol;Acc:HGNC:25565]
## ENSG00000000971 complement factor H [Source:HGNC Symbol;Acc:HGNC:4883]
## ENSG00000001036 fucosidase, alpha-L- 2, plasma [Source:HGNC Symbol;Acc:HGNC:4008]
Read in annotation package for homo sapiens (humans)
library('AnnotationDbi')
##
## Attaching package: 'AnnotationDbi'
## The following object is masked from 'package:dplyr':
##
## select
library('org.Hs.eg.db')
##
columns(org.Hs.eg.db)
## [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT"
## [5] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE"
## [9] "EVIDENCEALL" "GENENAME" "GO" "GOALL"
## [13] "IPI" "MAP" "OMIM" "ONTOLOGY"
## [17] "ONTOLOGYALL" "PATH" "PFAM" "PMID"
## [21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG"
## [25] "UNIGENE" "UNIPROT"
Add information to the counts data frame…
mean.counts$symbol <- mapIds(org.Hs.eg.db, keytype = 'ENSEMBL',
keys = row.names(mean.counts),
column = 'SYMBOL',
multiVals = 'first')
## 'select()' returned 1:many mapping between keys and columns
mean.counts$entrez <- mapIds(org.Hs.eg.db, keytype = 'ENSEMBL',
keys = row.names(mean.counts),
column = 'ENTREZID',
multiVals = 'first')
## 'select()' returned 1:many mapping between keys and columns
mean.counts$uniprot <- mapIds(org.Hs.eg.db, keytype = 'ENSEMBL',
keys = row.names(mean.counts),
column = 'UNIPROT',
multiVals = 'first')
## 'select()' returned 1:many mapping between keys and columns
head(mean.counts)
## control.means treated.means log2foldchange symbol entrez
## ENSG00000000003 900.75 658.00 -0.45303916 TSPAN6 7105
## ENSG00000000419 520.50 546.00 0.06900279 DPM1 8813
## ENSG00000000457 339.75 316.50 -0.10226805 SCYL3 57147
## ENSG00000000460 97.25 78.75 -0.30441833 C1orf112 55732
## ENSG00000000971 5219.00 6687.50 0.35769358 CFH 3075
## ENSG00000001036 2327.00 1785.75 -0.38194109 FUCA2 2519
## uniprot
## ENSG00000000003 A0A024RCI0
## ENSG00000000419 O60762
## ENSG00000000457 Q8IZE3
## ENSG00000000460 A0A024R922
## ENSG00000000971 A0A024R962
## ENSG00000001036 Q9BTY2
Q12:
head(mean.counts[row.names(up.regulated),])
## control.means treated.means log2foldchange symbol entrez
## ENSG00000004799 270.50 1429.25 2.401558 PDK4 5166
## ENSG00000006788 2.75 19.75 2.844349 MYH13 8735
## ENSG00000008438 0.50 2.75 2.459432 PGLYRP1 8993
## ENSG00000011677 0.50 2.25 2.169925 GABRA3 2556
## ENSG00000015413 0.50 3.00 2.584963 DPEP1 1800
## ENSG00000015592 0.50 2.25 2.169925 STMN4 81551
## uniprot
## ENSG00000004799 A4D1H4
## ENSG00000006788 Q9UKX3
## ENSG00000008438 O75594
## ENSG00000011677 P34903
## ENSG00000015413 A0A140VJI3
## ENSG00000015592 Q9H169
head(mean.counts[row.names(down.regulated),])
## control.means treated.means log2foldchange symbol entrez
## ENSG00000015520 32.00 6.00 -2.415037 NPC1L1 29881
## ENSG00000019186 26.50 1.75 -3.920566 CYP24A1 1591
## ENSG00000025423 295.00 54.25 -2.443020 HSD17B6 8630
## ENSG00000028277 88.25 22.00 -2.004093 POU2F2 5452
## ENSG00000029559 1.25 0.25 -2.321928 IBSP 3381
## ENSG00000049246 405.00 93.00 -2.122619 PER3 8863
## uniprot
## ENSG00000015520 A0A0C4DFX6
## ENSG00000019186 Q07973
## ENSG00000025423 A0A024RB43
## ENSG00000028277 P09086
## ENSG00000029559 P21815
## ENSG00000049246 A0A087WV69
I would not trust these results because we don’t know if these results are true or just false positives.
DESeq2 analysis
counts <- counts %>% rownames_to_column('ensgene')
dds <- DESeqDataSetFromMatrix(countData = counts,colData = metadata, design=~dex,
tidy = TRUE)
## converting counts to integer mode
## Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
## design formula are characters, converting to factors
dds
## class: DESeqDataSet
## dim: 38694 8
## metadata(1): version
## assays(1): counts
## rownames(38694): ENSG00000000003 ENSG00000000005 ...
## ENSG00000283120 ENSG00000283123
## rowData names(0):
## colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
## colData names(4): id dex celltype geo_id
dds <- DESeq(dds)
## estimating size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
Get the results for threshold of log2foldchange of +/- 2, alpha level of 0.05…
res <- results(dds)
head(as.data.frame(res))
## baseMean log2FoldChange lfcSE stat pvalue
## ENSG00000000003 747.1941954 -0.35070302 0.1682457 -2.0844697 0.03711747
## ENSG00000000005 0.0000000 NA NA NA NA
## ENSG00000000419 520.1341601 0.20610777 0.1010592 2.0394752 0.04140263
## ENSG00000000457 322.6648439 0.02452695 0.1451451 0.1689823 0.86581056
## ENSG00000000460 87.6826252 -0.14714205 0.2570073 -0.5725210 0.56696907
## ENSG00000000938 0.3191666 -1.73228897 3.4936010 -0.4958463 0.62000288
## padj
## ENSG00000000003 0.1630348
## ENSG00000000005 NA
## ENSG00000000419 0.1760317
## ENSG00000000457 0.9616942
## ENSG00000000460 0.8158486
## ENSG00000000938 NA
summary(res)
##
## out of 25258 with nonzero total read count
## adjusted p-value < 0.1
## LFC > 0 (up) : 1563, 6.2%
## LFC < 0 (down) : 1188, 4.7%
## outliers [1] : 142, 0.56%
## low counts [2] : 9971, 39%
## (mean count < 10)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?results
Significance of 0.01…
resSig01 <- results(dds, alpha = .01)
#head(as.data.frame(resSig01))
summary(resSig01)
##
## out of 25258 with nonzero total read count
## adjusted p-value < 0.01
## LFC > 0 (up) : 850, 3.4%
## LFC < 0 (down) : 581, 2.3%
## outliers [1] : 142, 0.56%
## low counts [2] : 9033, 36%
## (mean count < 6)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?results
Visualize
plot( res$log2FoldChange, -log(res$padj),
xlab="Log2(FoldChange)",
ylab="-Log(P-value)")
abline(h = -log(0.05), col = 'red')
abline(v = log(2, base = 2))
abline(v = -log(2, base = 2))
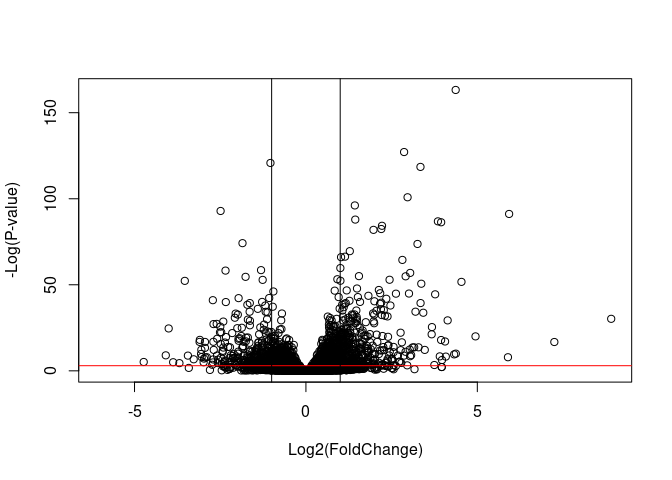
mycols <- rep("gray", nrow(res))
mycols[ abs(res$log2FoldChange) > 2 ] <- "red"
inds <- (res$padj < 0.01) & (abs(res$log2FoldChange) > 2 )
mycols[ inds ] <- "blue"
# Volcano plot with custom colors
plot( res$log2FoldChange, -log(res$padj),
col=mycols, ylab="-Log(P-value)", xlab="Log2(FoldChange)" )
# Cut-off lines
abline(v=c(-2,2), col="gray", lty=2)
abline(h=-log(0.1), col="gray", lty=2)
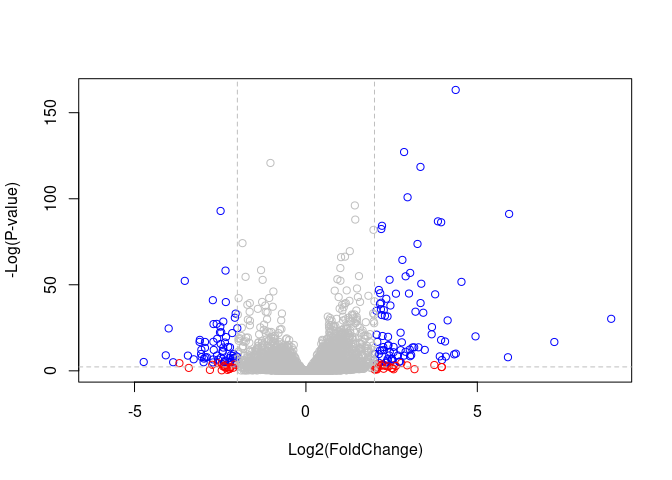
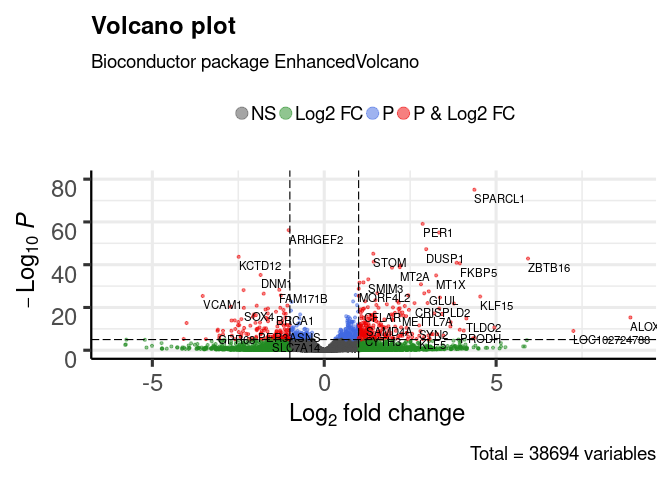
We can use the enhanced volcano plotting functions in bioconductor. First, let’s add the annotations.
x <- as.data.frame(res)
x$symbol <- mapIds(org.Hs.eg.db,
keys=row.names(x),
keytype="ENSEMBL",
column="SYMBOL",
multiVals="first")
## 'select()' returned 1:many mapping between keys and columns
library(EnhancedVolcano)
## Loading required package: ggplot2
## Loading required package: ggrepel
EnhancedVolcano(x, lab = x$symbol, x = 'log2FoldChange', y = 'pvalue')