BGGN 213 Spring 2019 Classwork
https://androidpcguy.github.io/bggn213/
Class 17 - Network Analysis
Akshara Balachandra 5/29/2019
Set up Cytoscape and R connection
Load the required packages RCy3 from bioconductor and igraph from CRAN.
library(RCy3)
## Registered S3 method overwritten by 'R.oo':
## method from
## throw.default R.methodsS3
library(igraph)
##
## Attaching package: 'igraph'
## The following objects are masked from 'package:stats':
##
## decompose, spectrum
## The following object is masked from 'package:base':
##
## union
Check connection to Cytoscape…
cytoscapePing()
## [1] "You are connected to Cytoscape!"
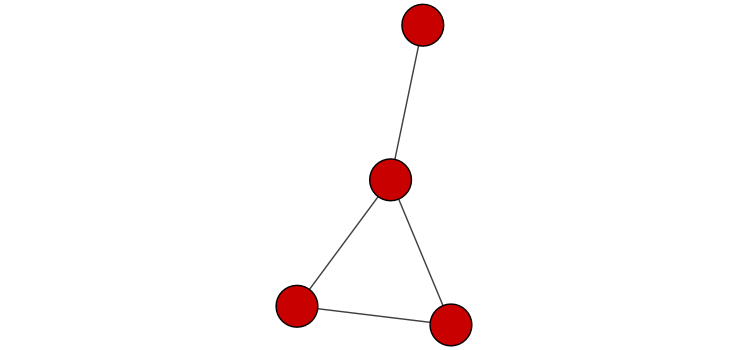
Make a simple graph
g <- makeSimpleIgraph()
createNetworkFromIgraph(g,"myGraph")
## Loading data...
## Applying default style...
## Applying preferred layout...
## networkSUID
## 7763
Include the graph in this report.
fig <- exportImage(filename="demo", type="png", height=350)
## Warning: This file already exists. A Cytoscape popup
## will be generated to confirm overwrite.
knitr::include_graphics('demo.png')
Change the visual style…
setVisualStyle("Marquee")
## message
## "Visual Style applied."
fig <- exportImage(filename="demo_marquee", type="png", height=350)
## Warning: This file already exists. A Cytoscape popup
## will be generated to confirm overwrite.
knitr::include_graphics("./demo_marquee.png")
Actual metagenomics data
Read the data….
prok_vir_cor <- read.delim("./data/virus_prok_cor_abundant.tsv", stringsAsFactors = FALSE)
## Have a peak at the first 6 rows
head(prok_vir_cor)
## Var1 Var2 weight
## 1 ph_1061 AACY020068177 0.8555342
## 2 ph_1258 AACY020207233 0.8055750
## 3 ph_3164 AACY020207233 0.8122517
## 4 ph_1033 AACY020255495 0.8487498
## 5 ph_10996 AACY020255495 0.8734617
## 6 ph_11038 AACY020255495 0.8740782
Create the graph
g <- graph.data.frame(prok_vir_cor, directed = F)
class(g)
## [1] "igraph"
g
## IGRAPH ede583f UNW- 845 1544 --
## + attr: name (v/c), weight (e/n)
## + edges from ede583f (vertex names):
## [1] ph_1061 --AACY020068177 ph_1258 --AACY020207233
## [3] ph_3164 --AACY020207233 ph_1033 --AACY020255495
## [5] ph_10996--AACY020255495 ph_11038--AACY020255495
## [7] ph_11040--AACY020255495 ph_11048--AACY020255495
## [9] ph_11096--AACY020255495 ph_1113 --AACY020255495
## [11] ph_1208 --AACY020255495 ph_13207--AACY020255495
## [13] ph_1346 --AACY020255495 ph_14679--AACY020255495
## [15] ph_1572 --AACY020255495 ph_16045--AACY020255495
## + ... omitted several edges
Plot it
plot(g, vertex.label = NA)

Make the nodes smaller
plot(g, vertex.label = NA, vertex.size = 3)

Network queries
V(g)
## + 845/845 vertices, named, from ede583f:
## [1] ph_1061 ph_1258 ph_3164 ph_1033 ph_10996
## [6] ph_11038 ph_11040 ph_11048 ph_11096 ph_1113
## [11] ph_1208 ph_13207 ph_1346 ph_14679 ph_1572
## [16] ph_16045 ph_1909 ph_1918 ph_19894 ph_2117
## [21] ph_2231 ph_2363 ph_276 ph_2775 ph_2798
## [26] ph_3217 ph_3336 ph_3493 ph_3541 ph_3892
## [31] ph_4194 ph_4602 ph_4678 ph_484 ph_4993
## [36] ph_4999 ph_5001 ph_5010 ph_5286 ph_5287
## [41] ph_5302 ph_5321 ph_5643 ph_6441 ph_654
## [46] ph_6954 ph_7389 ph_7920 ph_8039 ph_8695
## + ... omitted several vertices
E(g)
## + 1544/1544 edges from ede583f (vertex names):
## [1] ph_1061 --AACY020068177 ph_1258 --AACY020207233
## [3] ph_3164 --AACY020207233 ph_1033 --AACY020255495
## [5] ph_10996--AACY020255495 ph_11038--AACY020255495
## [7] ph_11040--AACY020255495 ph_11048--AACY020255495
## [9] ph_11096--AACY020255495 ph_1113 --AACY020255495
## [11] ph_1208 --AACY020255495 ph_13207--AACY020255495
## [13] ph_1346 --AACY020255495 ph_14679--AACY020255495
## [15] ph_1572 --AACY020255495 ph_16045--AACY020255495
## [17] ph_1909 --AACY020255495 ph_1918 --AACY020255495
## [19] ph_19894--AACY020255495 ph_2117 --AACY020255495
## + ... omitted several edges
Calculate some graph theory measures…
cb <- cluster_edge_betweenness(g)
## Warning in cluster_edge_betweenness(g): At community.c:460 :Membership
## vector will be selected based on the lowest modularity score.
## Warning in cluster_edge_betweenness(g): At community.c:467 :Modularity
## calculation with weighted edge betweenness community detection might not
## make sense -- modularity treats edge weights as similarities while edge
## betwenness treats them as distances
plot(cb, y = g, vertex.label = NA, vertex.size = 3)
Extract membership information
head(membership(cb))
## ph_1061 ph_1258 ph_3164 ph_1033 ph_10996 ph_11038
## 1 2 3 4 4 4
Node degree
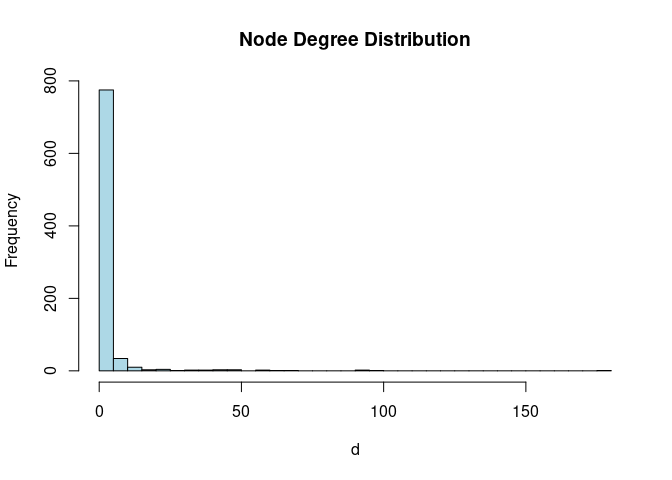
# Calculate and plot node degree of our network
d <- degree(g)
hist(d, breaks=30, col="lightblue", main ="Node Degree Distribution")
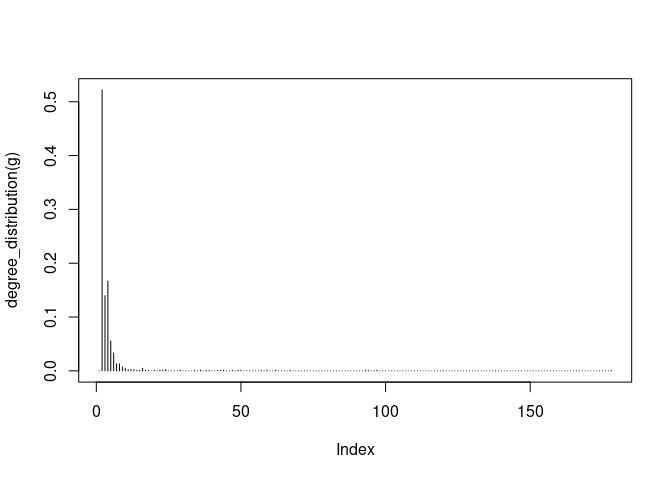
plot( degree_distribution(g), type="h" )
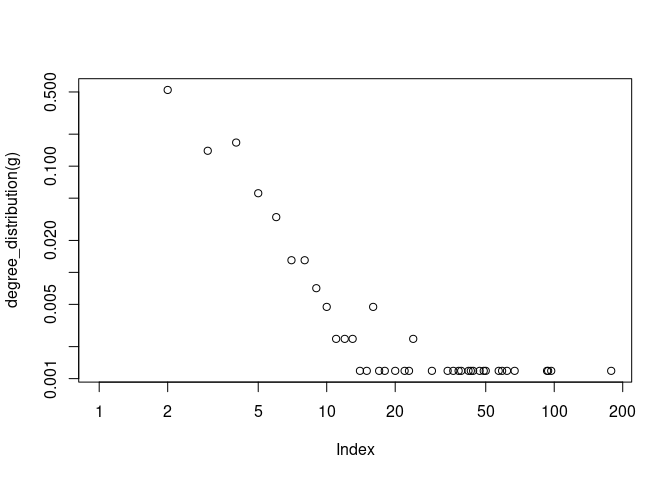
plot(degree_distribution(g), log = 'xy')
## Warning in xy.coords(x, y, xlabel, ylabel, log): 138 y values <= 0 omitted
## from logarithmic plot
Centrality Analaysis
Answer this question: Which nodes are the most important and why???
First method: PageRank
pr <- page_rank(g)
head(pr$vector)
## ph_1061 ph_1258 ph_3164 ph_1033 ph_10996
## 0.0011834320 0.0011599483 0.0019042088 0.0005788564 0.0005769663
## ph_11038
## 0.0005745460
Plot graph with nodes scaled by page rank score
# Make a size vector btwn 2 and 20 for node plotting size
v.size <- BBmisc::normalize(pr$vector, range=c(2,20), method="range")
plot(g, vertex.size=v.size, vertex.label=NA)

Second method: node degree
v.size <- BBmisc::normalize(d, range=c(2,20), method="range")
plot(g, vertex.size=v.size, vertex.label=NA)

Third method: betweenness
b <- betweenness(g)
v.size <- BBmisc::normalize(b, range=c(2,20), method="range")
plot(g, vertex.size=v.size, vertex.label=NA)

Read taxonomic classification for network annotation
phage_id_affiliation <- read.delim("./data/phage_ids_with_affiliation.tsv")
head(phage_id_affiliation)
## first_sheet.Phage_id first_sheet.Phage_id_network phage_affiliation
## 1 109DCM_115804 ph_775 <NA>
## 2 109DCM_115804 ph_775 <NA>
## 3 109DCM_115804 ph_775 <NA>
## 4 109DCM_115804 ph_775 <NA>
## 5 109DCM_115804 ph_775 <NA>
## 6 109DCM_115804 ph_775 <NA>
## Domain DNA_or_RNA Tax_order Tax_subfamily Tax_family Tax_genus
## 1 <NA> <NA> <NA> <NA> <NA> <NA>
## 2 <NA> <NA> <NA> <NA> <NA> <NA>
## 3 <NA> <NA> <NA> <NA> <NA> <NA>
## 4 <NA> <NA> <NA> <NA> <NA> <NA>
## 5 <NA> <NA> <NA> <NA> <NA> <NA>
## 6 <NA> <NA> <NA> <NA> <NA> <NA>
## Tax_species
## 1 <NA>
## 2 <NA>
## 3 <NA>
## 4 <NA>
## 5 <NA>
## 6 <NA>
bac_id_affi <- read.delim("./data/prok_tax_from_silva.tsv", stringsAsFactors = FALSE)
head(bac_id_affi)
## Accession_ID Kingdom Phylum Class Order
## 1 AACY020068177 Bacteria Chloroflexi SAR202 clade marine metagenome
## 2 AACY020125842 Archaea Euryarchaeota Thermoplasmata Thermoplasmatales
## 3 AACY020187844 Archaea Euryarchaeota Thermoplasmata Thermoplasmatales
## 4 AACY020105546 Bacteria Actinobacteria Actinobacteria PeM15
## 5 AACY020281370 Archaea Euryarchaeota Thermoplasmata Thermoplasmatales
## 6 AACY020147130 Archaea Euryarchaeota Thermoplasmata Thermoplasmatales
## Family Genus Species
## 1 <NA> <NA> <NA>
## 2 Marine Group II marine metagenome <NA>
## 3 Marine Group II marine metagenome <NA>
## 4 marine metagenome <NA> <NA>
## 5 Marine Group II marine metagenome <NA>
## 6 Marine Group II marine metagenome <NA>
## Extract out our vertex names
genenet.nodes <- as.data.frame(vertex.attributes(g), stringsAsFactors=FALSE)
head(genenet.nodes)
## name
## 1 ph_1061
## 2 ph_1258
## 3 ph_3164
## 4 ph_1033
## 5 ph_10996
## 6 ph_11038
How many phage entries do we have?
sum(grepl('ph_', genenet.nodes$name))
## [1] 764
Merge annotation data with phylogenetic data…
# We dont need all annotation data so lets make a reduced table 'z' for merging
z <- bac_id_affi[,c("Accession_ID", "Kingdom", "Phylum", "Class")]
n <- merge(genenet.nodes, z, by.x="name", by.y="Accession_ID", all.x=TRUE)
head(n)
## name Kingdom Phylum Class
## 1 AACY020068177 Bacteria Chloroflexi SAR202 clade
## 2 AACY020207233 Bacteria Deferribacteres Deferribacteres
## 3 AACY020255495 Bacteria Proteobacteria Gammaproteobacteria
## 4 AACY020288370 Bacteria Actinobacteria Acidimicrobiia
## 5 AACY020396101 Bacteria Actinobacteria Acidimicrobiia
## 6 AACY020398456 Bacteria Proteobacteria Gammaproteobacteria
# Again we only need a subset of `phage_id_affiliation` for our purposes
y <- phage_id_affiliation[, c("first_sheet.Phage_id_network", "phage_affiliation","Tax_order", "Tax_subfamily")]
# Add the little phage annotation that we have
x <- merge(x=n, y=y, by.x="name", by.y="first_sheet.Phage_id_network", all.x=TRUE)
## Remove duplicates from multiple matches
x <- x[!duplicated( (x$name) ),]
head(x)
## name Kingdom Phylum Class
## 1 AACY020068177 Bacteria Chloroflexi SAR202 clade
## 2 AACY020207233 Bacteria Deferribacteres Deferribacteres
## 3 AACY020255495 Bacteria Proteobacteria Gammaproteobacteria
## 4 AACY020288370 Bacteria Actinobacteria Acidimicrobiia
## 5 AACY020396101 Bacteria Actinobacteria Acidimicrobiia
## 6 AACY020398456 Bacteria Proteobacteria Gammaproteobacteria
## phage_affiliation Tax_order Tax_subfamily
## 1 <NA> <NA> <NA>
## 2 <NA> <NA> <NA>
## 3 <NA> <NA> <NA>
## 4 <NA> <NA> <NA>
## 5 <NA> <NA> <NA>
## 6 <NA> <NA> <NA>
Save the annotation data to the graph nodes..
genenet.nodes <- x
Send network to Cytoscape using RCy3
# delete any existing networks
deleteAllNetworks()
# Set the main nodes colname to the required "id"
colnames(genenet.nodes)[1] <- "id"
Add the network data to edges/nodes and send to cytoscape
genenet.edges <- data.frame(igraph::as_edgelist(g))
# Set the main edges colname to the required "source" and "target"
colnames(genenet.edges) <- c("source","target")
# Add the weight from igraph to a new column...
genenet.edges$Weight <- igraph::edge_attr(g)$weight
# Send as a new network to Cytoscape
createNetworkFromDataFrames(genenet.nodes,genenet.edges,
title="Tara_Oceans")
## Loading data...
## Applying default style...
## Applying preferred layout...
## networkSUID
## 7807
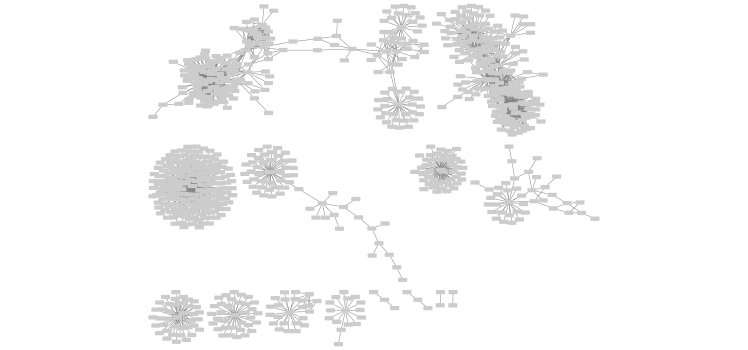
Save a figure of the cytoscape network here…
fig <- exportImage(filename="tara_oceans", type="png", height=350)
## Warning: This file already exists. A Cytoscape popup
## will be generated to confirm overwrite.
knitr::include_graphics("./tara_oceans.png")